
Peto's Aging
How our lifespans may be an evolutionary accident

Consider this:
An elephant has over a thousand times more cells than a human.
Cells accrue mutations independently.
Cancer is mostly driven by mutations.
So why do we not get cancer a thousand times more frequently than mice?
This is Peto’s Paradox. There are many proposed “answers” to this paradox, but there’s a major clue found when investigating the number of mutations animals acquire per year. There seems to be a natural limit to the number of mutations each animal acquires per cell on average, and that number is surprisingly small:
3,000.
And it takes mice a little over three years to accumulate these 3,000 mutations, compared to sixty years for an elephant and around eighty for a human. Take a look at the graph below comparing mutation rate per year with an animal’s mass:
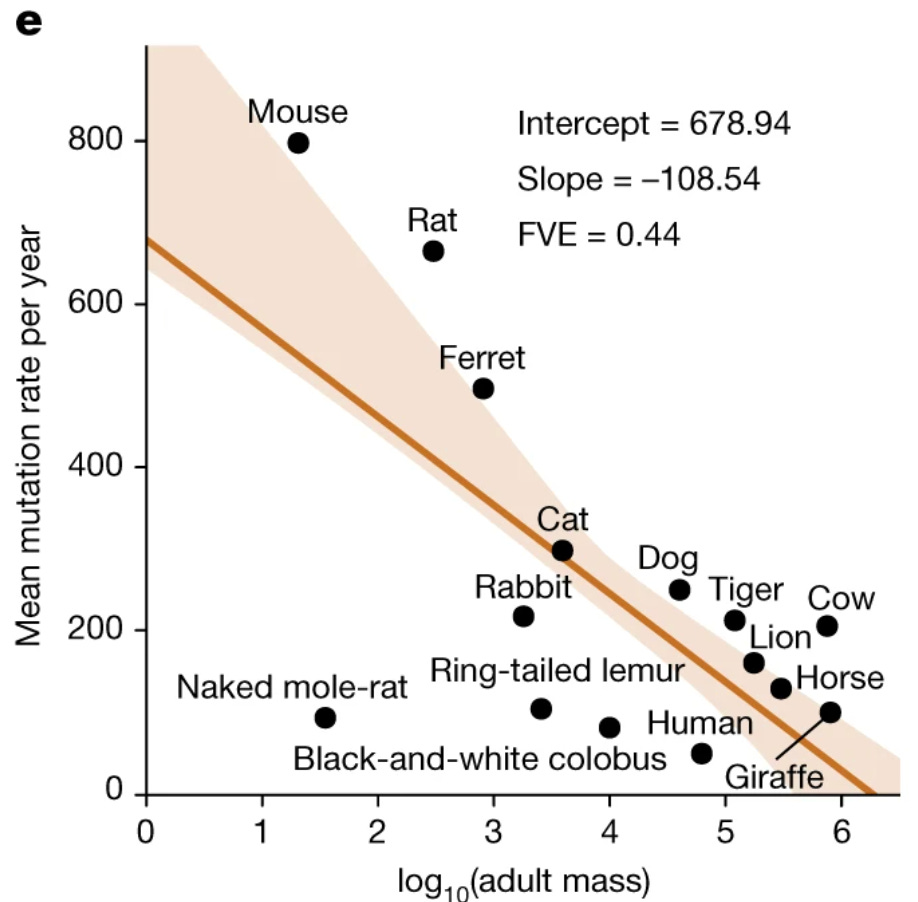
from Cagan et al. 2022
This creates a straightforward explanation – larger animals, needing to avoid dying at a young age due to cancer – evolved better mechanisms to protect and repair DNA. This is not the entire story, as better immune surveillance and differences in metabolism contribute as well, but it’s as close to a complete picture as you get in biology.
Entirely separately, longevity researchers have observed a correlation between the size of an animal and its maximum lifespan as well1:
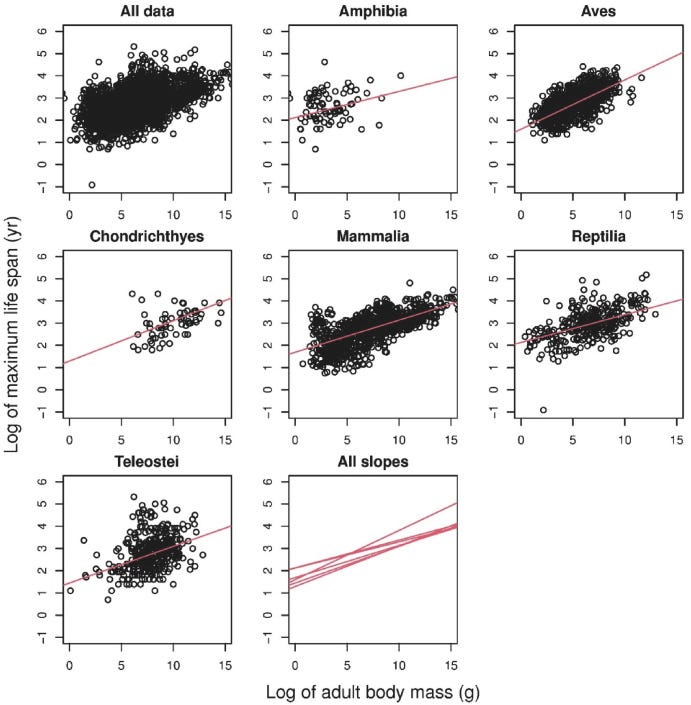
Why? It’s not entirely clear. The aging field has assumed that this has something to do with metabolism, as larger animals tend to better pace their energy uptake. But there’s another aspect to the story, in that there’s a strong association between mutational burden and lifespan that dwarfs both the association between mass and mutations and the association between mass and lifespan:
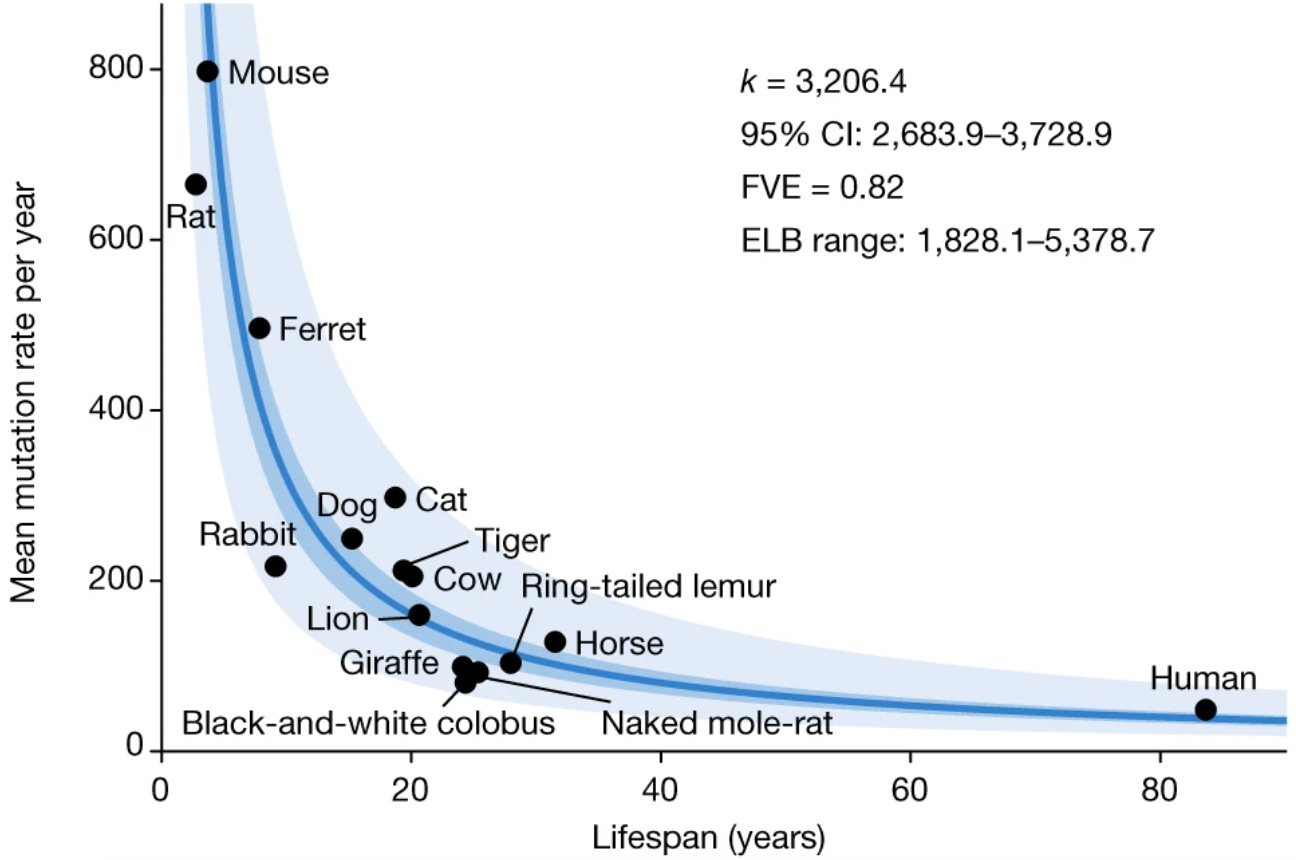
from Cagan et al. 2022, again
In fact, if you account for the association between mutational burden and lifespan, the original link between body mass and lifespan ceases to be significant.
So there are three interesting observations (drawn below):

There is a strong association between larger body mass and improved ability to maintain the genome. This is likely to be an evolutionarily-driven adaptation for survival.
There is a moderate association between larger body mass and longer lifespan.
There is a very strong association between improved ability to maintain the genome and maximum lifespan. This is unlikely to be evolutionarily-driven, as aging is something that tends to occur after an organism’s reproductive window.
A unifying explanation: Extended maximum lifespan in larger animals is an evolutionary accident of requiring better genome stability mechanisms to avoid cancer. Maximum lifespan is a byproduct of solving Peto’s Paradox. If you stop the things that cause you to get cancer, you get extended life for free. Preventing damage to the genome is a path to living longer.
This isn’t the only reason to view genomic instability as a, if not the, major driver of aging. Effectively every single disease in humans that resembles a form of accelerated aging is caused by a mutation in a gene related to genomic stability:
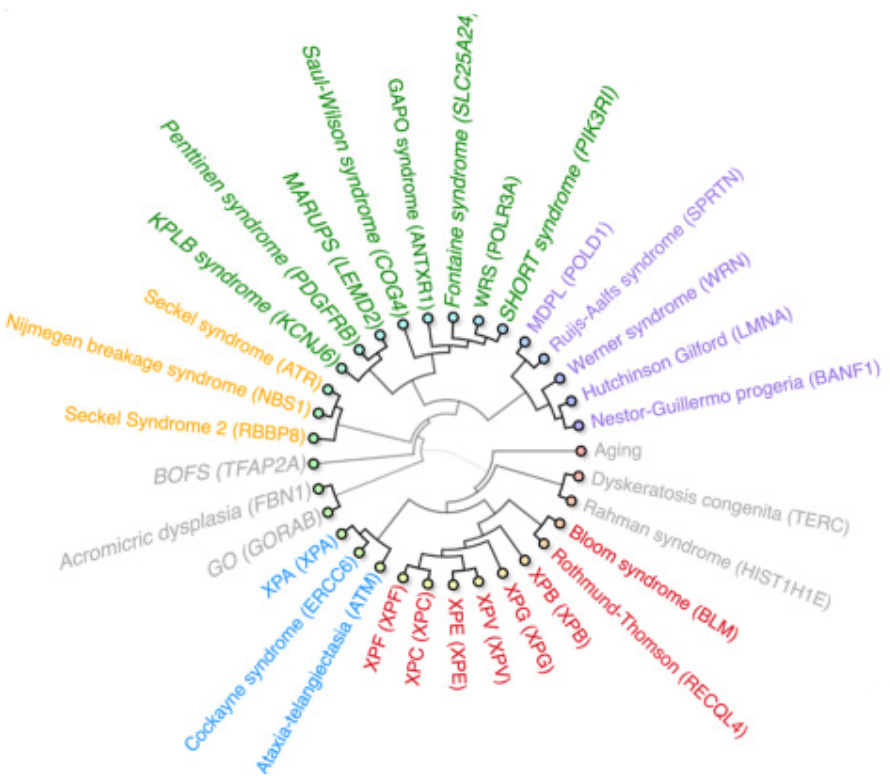
from Worm et al. 2024
The age of menopause in women, which is itself linked to mortality and morbidity, is strongly tied to DNA repair genes. A better version of a DNA maintenance protein is heavily enriched in centenarians. Molecular markers immediately downstream of genomic damage - DNA methylation, mutations - change on a constant basis with age, suggesting they do not participate in the secondary feedback loops that define every other aging hallmark.
Note I emphasize lifespan here instead of healthspan. Healthspan is linked to a separate suite of programs. But that is a story for a different time.
So aging is caused by mutations, then?
No.
There is a rare disease (a mutation in POLE) that leads to an accumulation of mutations across the genome at a frequency up to ten times higher than in the general human population. Unsurprisingly, this leads to a high risk of cancer. Perhaps surprisingly, there does not seem to be much of an effect on aging. The increase is not tied to any particular cell type, either:
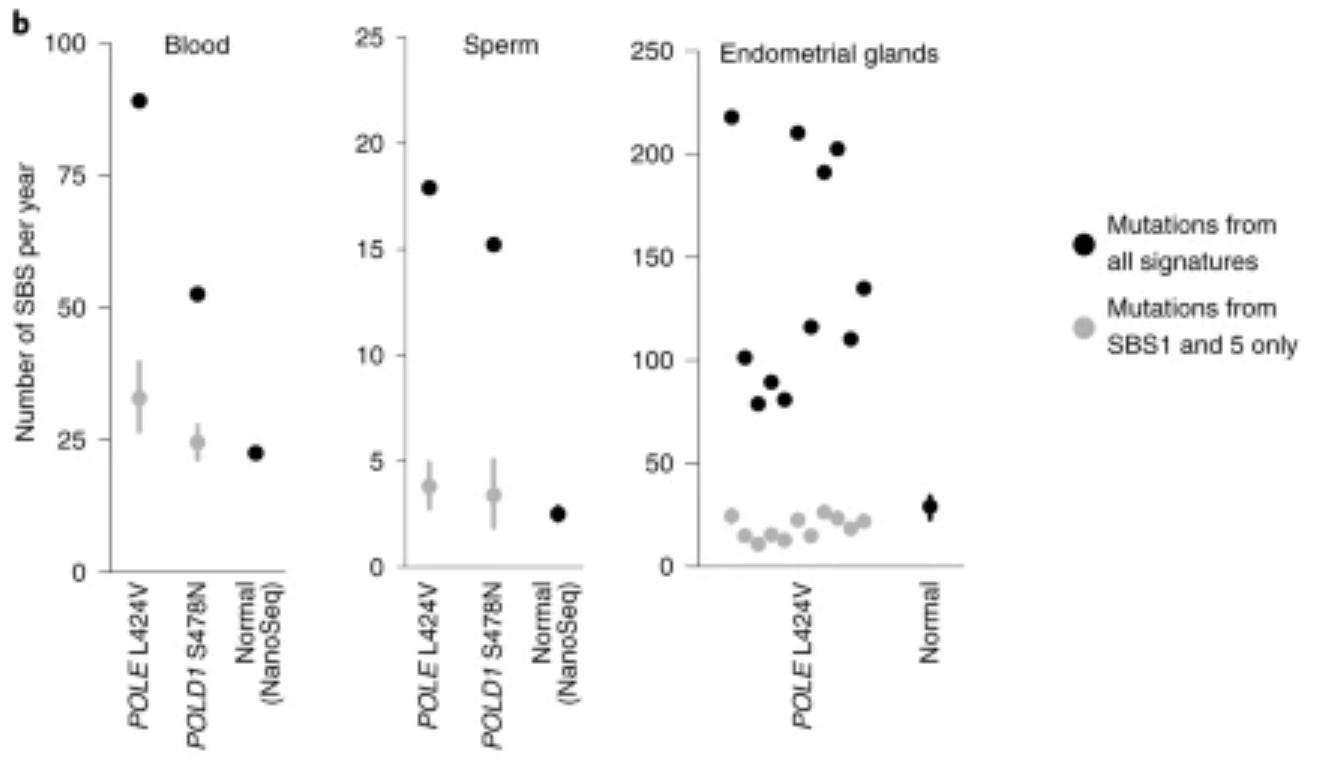
from Robinson et al. 2021
It’s also unlikely to be mutations in the mitochondria, as mice have been generated with 500-fold more mitochondrial mutations while not aging a whole lot faster.
To be clear, mutations have been linked to manifestations of aging. The most well-known example is clonal hematopoiesis, where blood stem cells pick up mutations that make them better at dividing which then leads to an increased rate of cancer. There are also a number of studies emerging linking clonal expansion of various cell types in the brain with neurological disorders such as Parkinson’s and Alzheimer’s. The effect sizes for these studies tend to be pretty weak, however. In some cases, mutation-induced clonal expansion has actually been shown to have positive effects, such as by improving outcomes for some cancers.
So what is it?
Here’s a pretty simple model:
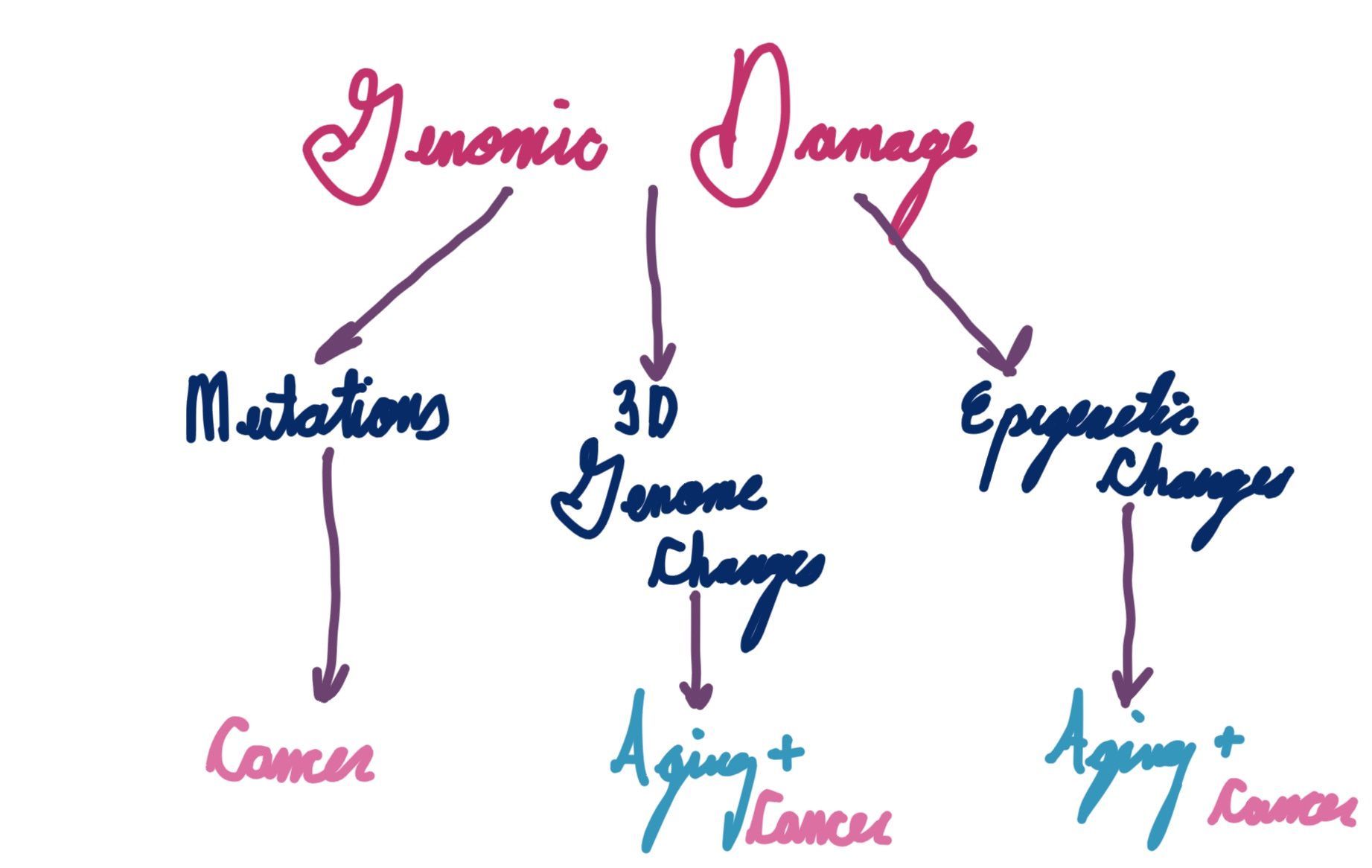
I drew this on an iPad on a plane undergoing turbulence
I’m being quite vague here, but this is intentional. The jury is still out on which aspects of genome instability are the most important for aging. David Sinclair’s information theory, likely one of the most well known theories of aging, centers around epigenetics (particularly histones) as the key determinant. 3D genome structure, thus far under-appreciated due to the difficulty of studying it, may play a pivotal role as well. Cell damage signaling is important too, though it may be more tied to healthspan rather than lifespan.
It could also depend on the cell type. About a year ago, I embarked on a quest to map every single disease linked to DNA repair in humans and whether it resembled aging or not. The diagram below is a little inaccurate, but the conclusion I came to was that repair pathways that predominantly impact slow or non-dividing cells are linked to aging, while pathways that impact fast-dividing cells lead to a higher risk of cancer.

Circling back to the main point - if it is true that maximum lifespan is an evolutionary accident related to large animals improving genome stability to avoid dying of cancer, what are the implications?
Two immediately come to mind.
Implication 1: Focus on preventing or reversing genome instability
On the reversal front, the field of epigenetic reprogramming has gained an enormous amount of attention. Other authors have written (one) (two) excellent posts on the subject, so I will leave the reader to peruse those references.
On the prevention side, however, there is remarkably little work being done2. This is surprising, because it seems like a much easier angle (an ounce of prevention is worth a pound of cure, as they say), and because there’s a lot of low hanging fruit. Spermidine, a readily available supplement, has been shown to enhance genome stability (side note: sperm accumulate fewer mutations than other cell types, despite being quite proliferative. spermidine may be why). Deep sea vent bacteria make compounds that protect their DNA. Bowhead whales, which live well past 200 years, have a better version of a key DNA repair enzyme that could have translational potential in humans.
Implication 2: Figure out which pathways drive cancer prevention, and which drive longer lifespan
You may have heard that elephants have many copies of p53, the “guardian of the genome” gene. What you may not know is that mice that have been engineered to have more p53 copies have reduced lifespan and poorer health.
Not every pathway that improves cancer resistance leads to longer lifespan. In many cases, it’s the opposite. For genes related to the cell cycle - such as p53 - having too much “error checking” leads to a failure of stem cells to divide properly, leading to accelerated aging but less cancer. On the flip side, having longer telomeres can be beneficial for aging while also promoting cancer growth. You can imagine a diagram that looks like this:

Note that if the Peto’s Aging theory is correct, then most interventions in genome stability should be somewhere in the middle. This pans out with human data, as diseases resembling accelerated aging tend to come with a higher cancer risk as well. On the flip side, reducing calories both slows down cancer onset and is one of the few interventions that has been shown to extend lifespan. The same is true for rapamycin.
The implication here is pretty profound - if you can prevent cancer, in most cases you should be able to extend lifespan as well. The key will be to prove that this is true by identifying more interventions that can slow the onset of both cancer and death.

Interestingly, this is true only across species. The opposite is true within species. Larger dogs live shorter than smaller dogs. My suspicion is that evolving better DNA repair lags behind evolving faster metabolism.
Disclosure: I am working on this.